
应用领域介绍:
计算材料学是近年里飞速发展的一门新兴交叉学科。它综合了凝聚态物理、材料物理学、理论化学、材料力学和工程力学等多个相关学科。其目的是利用高性能计算机,模拟材料的各种物理化学性质,深入理解材料从微观到宏观多个尺度的各类现象与特征,并对于材料的结构和物性进行预测,从而达到设计新材料的目的。
解决方案与服务内容:
材料科学方向的计算机模拟服务,通过在新材料计算平台上部署丰富的科学计算软件,从微观跨越介观和宏观尺度,模拟材料研究中的各类问题和现象,并帮助用户实现通过计算来设计新材料的目的。
典型案例:
案例一:水的量子效应
北京大学量子材料科学中心江颖研究员课题组和王恩哥院士课题组以及物理学院李新征研究员、华中科技大学吕京涛研究员合作,国际上率先测定了氢键的量子成分,揭示了水的核量子效应,从全新的角度诠释了水的奥秘。相关研究成果于2016年4月15日刊发在国际顶级学术期刊《科学》(Science DOI: 10.1126/science.aaf2042)上。
“水的结构是什么”这是《科学》杂志在创刊125周年的特刊中提出的125个最具挑战性的科学问题之一。水的结构之所以如此复杂,其中一个很重要的原因就是源于水分子之间的氢键相互作用。人们通常认为氢键的本质为经典的静电相互作用,然而由于氢原子核质量很小,其量子特性(量子隧穿和量子涨落)往往不可忽视,因此氢键同时也包含一定的量子成分。氢核的量子效应对氢键相互作用到底有多大影响?或者说氢键的量子成分究竟有多大?这个问题对于理解水/冰的微观结构和反常物性至关重要。但是,氢核的量子化研究无论对于实验还是理论都非常具有挑战性。
为实现对氢核量子特性的精确探测和描述,江颖课题组和王恩哥课题组近年来在相关实验技术和理论方法上分别取得突破。他们成功发展了对于氢核敏感的超高分辨扫描探针显微术,开发了基于第一性原理的路径积分分子动力学方法(全量子化计算),实现了单个水分子内部自由度的成像和水的氢键网络构型的直接识别[Nature Materials 13, 184 (2014)和Nature Communications 5, 4056 (2014],并在此基础上探测到氢核的动态转移过程[Nature Physics 11, 235 (2015)]。
最近,他们又基于扫描隧道显微镜研发了一套“针尖增强的非弹性电子隧穿谱”技术,突破了传统非弹性电子隧穿谱技术在信噪比和分辨率方面的限制,国际上首次获得了单个水分子的高分辨振动谱,并由此测得了单个氢键的强度。通过可控的同位素替换实验,并结合全量子化计算模拟,他们发现氢键的量子成分可远大于室温下的热能,表明氢核的量子效应不只是对经典相互作用的简单修正,其足以对水的结构和性质产生显著的影响。进一步深入分析表明,氢核的非简谐零点运动会弱化弱氢键,强化强氢键,这个物理图像对于各种氢键体系具有相当的普适性,澄清了学术界长期争论的氢键的量子本质。
《科学》杂志的审稿人盛赞该工作是“实验的杰作(tour de force experiments)”、“一定会引起谱学界的广泛兴趣(they are certainly of interest to the spectroscopy community)”、“为研究氢核量子效应提供了一个绝佳的平台(this measurement is unique and provides a fantastic opportunity to examine the contribution of quantum motion of the proton)”。

水的量子效应
案例二:调控表面原子结构制备优异电催化剂:
控制催化剂表面原子结构是提高催化剂性能的有效途径。对于金属氧化物,表面电子结构可控制备及原子尺度结构与催化性能关系研究仍然具有巨大挑战。
采用动力学控制的气相离子交换策略,在高导电碳纤维基底上可控制备了长度及直接可控的CoO纳米线阵列。原子分辨率球差电镜实验及模拟分析表明CoO纳米线表面暴露织构化的金子塔结构,这种特殊的结构由两个{100}面和两个高表面能的{111}面组成。X射线近边吸收结果表明CoO纳米线表面富集氧空位。DFT计算表明在{111}表面氧空位的生长能比{100}面和{110}面低3 eV,因此这种特殊的金字塔结构表面易于富集氧空位。
上述特殊的表面原子结构使CoO纳米线阵列获得了优异的ORR/OER催化活性,其氧还原性能(ORR)的开启电压及ORR饱和电流已经逼近贵金属Pt;其氧析出性能(OER)已经优于商用RuO2催化剂;其ORR-OER双功能催化的ΔE=0.71 V是所有材料中最好的。
通过天河一号计算表明CoO纳米线优异的电催化性能与其表面原子结构密切相关,在金字塔{111}表面引入氧空位后能实现有效的反应物O2活化;在ORR/OER催化反应过程中,所有反应中间体获得最适合的吸附能变化;另外,在{111}表面引入氧空位后能在CoO能带中增加新的电子态,大大提高氧化物的导电性,实现电催化过程中快速的电荷转移。
该工作发表于Nature Communications, 2016, 7, 12876。
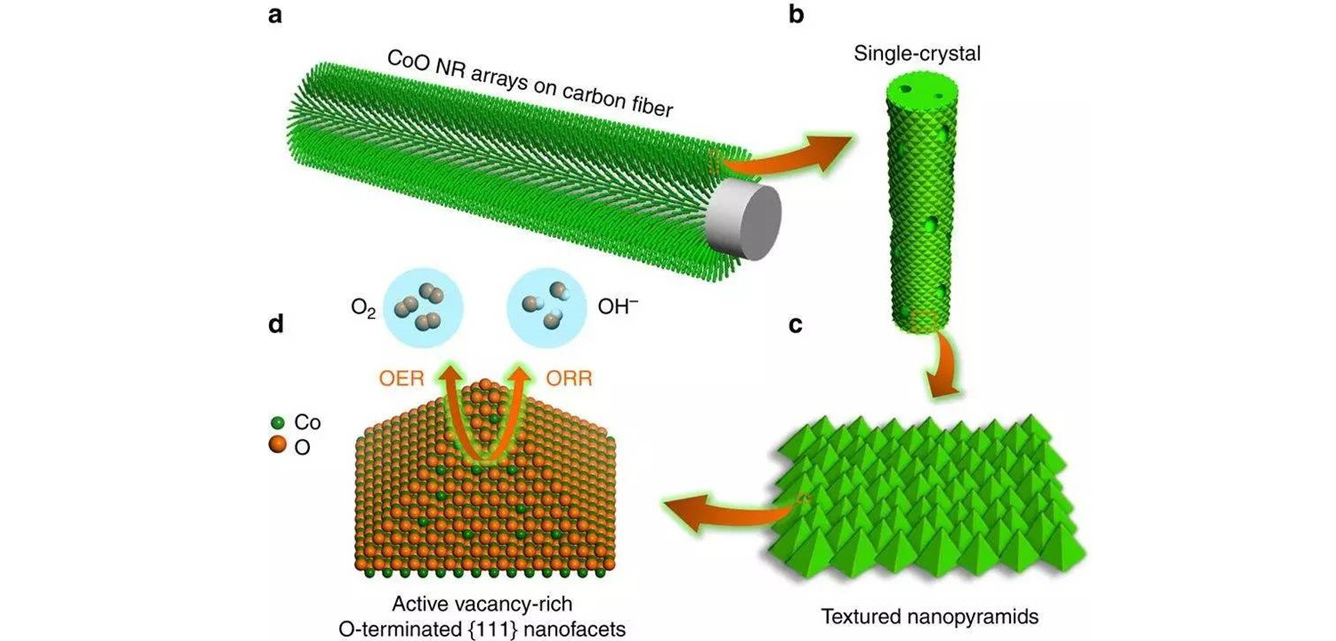
调控表面原子结构制备优异电催化剂
案例三:相变存储器研究
相变存储器在高速、海量存储等方面具有巨大潜力,已成为下一代非易失性存储技术的最佳解决方案。使用第一性理论计算能够从材料纳米尺度微观结构出发,模拟原子在外加热场、力场作用下的运动规律,弥补实验物理对微区现象表征时遇到的诸多困难,分析并预测其电学、光学特性,揭示材料相变的机制,对新型相变材料研发与工程化具有指导意义。
深圳大学通过理论计算,筛选出理想的掺杂元素Sc,经过实验测试ScSbTe相变材料,发现具有0.7 ns的相变速度和在皮秒脉冲作用下的105的循环操作寿命。
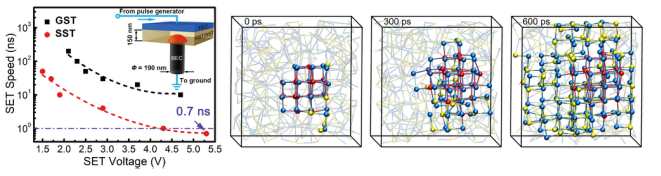
图 新型钪锑碲相变存储器件0.7纳秒的高速写入操作及其相变机理
通过分子动力学模拟,认为材料利用结构适配且更加稳定的钪碲化学键来加速晶核的孕育过程,显著降低形核过程的随机性,大幅加快结晶化即写入操作速度。
通过球差透射电子显微镜发现在TiSbTe材料中,Ti以两种形式存在:以TiTe2纳米片层结构紧邻Sb2Te3分布,和进入Sb2Te3晶格占据Sb位置。第一性原理计算与实验结果相符,同时我们认为TiTe2纳米片层结构以其低热导率而降低了材料的相变功耗,而进入Sb2Te3晶格占据Sb位置的Ti则可以提高材料的非晶化速度,进而提高相变速度。从原子尺寸的解释了TiSbTe材料的高速低功耗相变的原因。

图 TiSbTe晶格结构及其CDD计算结果
通过球差电镜观测并结合第一性原理计算,深圳大学提出了Sb2Te3的一个亚稳相:FCC相。通过分子动力学模拟得出了FCC相在非晶化过程中功耗更低的结论。
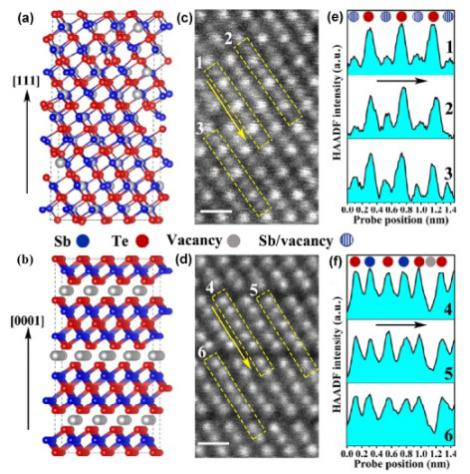
图 稳态和亚稳态Sb2Te3的第一性计算及球差电镜结果
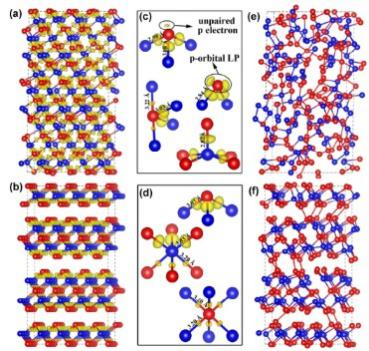
图 在相同的高温条件下FCC相需要更低的能量达到非晶化
通过第一性原理了计算并结合XPS计算结果得出了W掺杂后替代Sb进入Sb2Te3晶格。进而有利于细化晶粒,提高材料的使用寿命。

图 W掺杂有利于减小晶粒尺寸
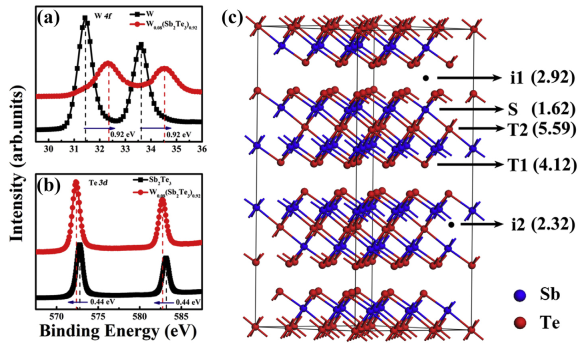
图 XPS结合形成能计算认为W占据Sb位置